

Migalastaté uma terapia oral inovadora desenvolvida para o tratamento da doença de Fabry, uma doença genética rara de armazenamento lisossômico que afeta múltiplos órgãos e reduz significativamente a qualidade de vida. Ao contrário das terapias tradicionais de substituição enzimática, o Migalastat atua estabilizando certas formas da própria enzima do corpo, permitindo-lhe funcionar de forma mais eficaz. Este artigo fornece uma visão geral completa e aprofundada do Migalastat, incluindo seu mecanismo de ação, aplicações clínicas, benefícios, limitações, adequação do paciente, perfil de segurança e impacto terapêutico no mundo real. Ele foi projetado para ajudar pacientes, cuidadores e partes interessadas na área da saúde a entender melhor como esta medicina de precisão está remodelando as estratégias de gerenciamento de doenças a longo prazo.
A doença de Fabry é uma doença hereditária rara de armazenamento lisossômico causada por mutações no gene GLA, que leva à atividade deficiente ou ausente da enzima alfa-galactosidase A (α-Gal A). Esta deficiência enzimática resulta no acúmulo de globotriaosilceramida (Gb3) em vários tecidos, incluindo vasos sanguíneos, rins, coração e sistema nervoso. Com o tempo, esse acúmulo leva a danos progressivos nos órgãos, dor crônica, insuficiência renal, doenças cardíacas e redução da expectativa de vida.
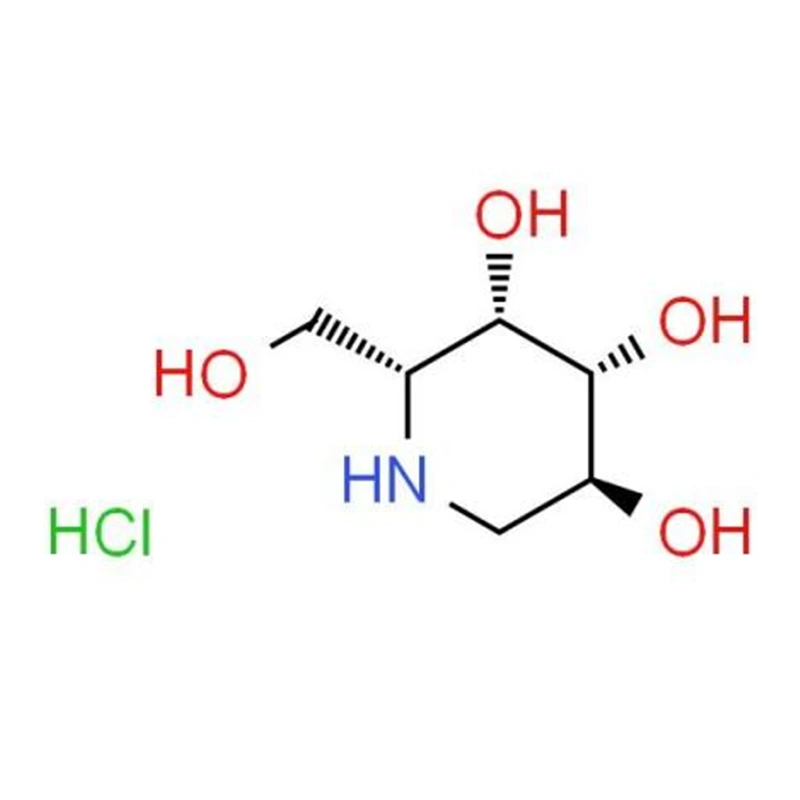
Migalastaté um acompanhante farmacológico oral desenvolvido como terapia direcionada para pacientes com doença de Fabry com mutações genéticas “aceitáveis” específicas. Em vez de substituir a enzima externamente, estabiliza a enzima do próprio paciente, ajudando-a a dobrar-se corretamente e a atingir os lisossomas, onde pode quebrar os substratos acumulados.
Este mecanismo representa um avanço significativo na medicina personalizada, oferecendo uma alternativa à terapia de reposição enzimática intravenosa (TRE) vitalícia.
O migalastat funciona como um inibidor competitivo reversível que se liga seletivamente a certas formas mal dobradas da alfa-galactosidase A. Essas mutações geralmente produzem enzimas que são estruturalmente instáveis, mas ainda potencialmente funcionais.
Seu mecanismo pode ser resumido em três etapas principais:
Esta abordagem de “acompanhante farmacológico” garante que as proteínas enzimáticas endógenas sejam resgatadas em vez de substituídas.
Resultado biológico chave:Redução do acúmulo de Gb3 e melhoria da função celular em múltiplos órgãos.
O Migalastat não é adequado para todos os pacientes com doença de Fabry. A sua eficácia depende se o paciente tem uma mutação geneticamente “aceitável”.
Os candidatos ideais normalmente incluem:
O teste genético é essencial antes de iniciar a terapia, pois apenas mutações específicas respondem eficazmente ao Migalastat.
O Migalastat é normalmente administrado como uma cápsula oral tomada em dias alternados. Deve ser tomado com o estômago vazio para otimizar a absorção.
| Parâmetro | Detalhes |
|---|---|
| Forma | Cápsula oral |
| Frequência de dosagem | Todos os dias |
| Condição de administração | Estômago vazio (2 horas antes ou depois da refeição) |
| Armazenar | Temperatura ambiente, local seco |
Os pacientes devem cumprir rigorosamente os esquemas de dosagem para manter efeitos consistentes de estabilização enzimática.
O Migalastat oferece várias vantagens importantes em comparação com as terapias tradicionais, particularmente para perfis genéticos adequados.
Esses benefícios melhoram significativamente a adesão e a qualidade de vida do paciente.
Apesar das vantagens, o Migalastat apresenta diversas limitações que devem ser consideradas.
Estas restrições destacam a importância do rastreio genético personalizado antes do início do tratamento.
O migalastat e a terapia de reposição enzimática (TRE) representam duas abordagens distintas para o manejo da doença de Fabry. A tabela a seguir descreve suas principais diferenças.
| Recurso | Migalastat | ERT (Terapia de Reposição Enzimática) |
|---|---|---|
| Via de administração | Cápsula oral | Infusão intravenosa |
| Freqüência | Todos os dias | A cada 2 semanas |
| Mecanismo | Acompanhante farmacológico | Substituição enzimática |
| Elegibilidade do paciente | Apenas mutações passíveis | Ampla aplicabilidade |
| Ambiente clínico | Em casa | Hospital/clínica |
Cada terapia tem seus pontos fortes e as decisões de tratamento dependem do perfil genético, da gravidade da doença e da preferência do paciente.
O Migalastat é geralmente bem tolerado, mas como todos os medicamentos, pode causar efeitos colaterais em alguns indivíduos.
Os efeitos colaterais comumente relatados incluem:
Efeitos adversos graves são raros, mas requerem atenção médica se ocorrerem. Recomenda-se a monitorização regular da função renal e cardíaca durante o tratamento.
A jornada do tratamento com o Migalastat começa com a confirmação genética de uma mutação passível de resposta. Assim que a elegibilidade for confirmada, os pacientes fazem a transição para um plano de tratamento estruturado.
O monitoramento contínuo garante que o tratamento permaneça eficaz e se ajuste à progressão da doença quando necessário.
Estudos clínicos demonstraram que o Migalastat pode melhorar ou estabilizar a função renal, reduzir a massa ventricular esquerda no coração e diminuir os biomarcadores da doença em pacientes adequados.
Estudos observacionais de longo prazo sugerem eficácia sustentada quando os pacientes mantêm adesão aos protocolos de tratamento. A investigação continua a avaliar o seu papel nas estratégias mais amplas de gestão da doença de Fabry.
Os ensaios clínicos em curso também exploram terapias combinadas e resultados de proteção de órgãos a longo prazo.
Q1: O Migalastat é uma cura para a doença de Fabry?
Não, o Migalastat não é uma cura. Ajuda a controlar os sintomas e retarda a progressão da doença em pacientes elegíveis.
P2: Com que rapidez o Migalastat funciona?
Melhorias bioquímicas podem ser observadas em meses, mas os benefícios clínicos podem demorar mais, dependendo da gravidade da doença.
Q3: As crianças podem usar o Migalastat?
As aprovações atuais concentram-se principalmente em pacientes adultos, embora estejam em andamento pesquisas para uso pediátrico.
Q4: O que acontece se uma dose for esquecida?
Os pacientes devem tomar a dose esquecida o mais rápido possível, a menos que esteja próximo da próxima dose programada.
Q5: O uso a longo prazo é seguro?
Estudos de longo prazo indicam segurança geralmente favorável, mas é necessária monitorização contínua.
O Migalastat representa um avanço significativo no tratamento da doença de Fabry, oferecendo uma opção terapêutica oral direcionada para pacientes com mutações genéticas específicas. Sua capacidade de estabilizar a função enzimática endógena proporciona uma abordagem única que difere fundamentalmente das terapias tradicionais de reposição enzimática.
À medida que a investigação avança, o Migalastat pode desempenhar um papel cada vez mais importante nas estratégias de medicina de precisão para doenças genéticas raras. O monitoramento clínico contínuo e a inovação irão refinar ainda mais suas aplicações e melhorar os resultados dos pacientes em longo prazo.
Com a crescente sensibilização e capacidades de rastreio genético, mais pacientes poderão ter acesso a esta terapia, transformando a forma como a doença de Fabry é gerida a nível mundial.
Para soluções farmacêuticas mais profissionais, colaboração em pesquisa e suporte para formulações avançadas relacionadas a terapias para doenças raras, entre em contato com:
Cosper Pharma Tech Co., Ltd.é especializada no desenvolvimento farmacêutico de alta qualidade e em tecnologias terapêuticas inovadoras projetadas para apoiar as necessidades globais de saúde.
Se você estiver interessado em oportunidades de parceria, dúvidas sobre produtos ou consultoria técnica, não hesite em nos contatar. Nossa equipe está pronta para apoiar seu projeto desde o desenvolvimento até a comercialização.
Contate-noshoje para explorar soluções farmacêuticas avançadas com a Cosper Pharma Tech Co., Ltd.
-OH")

-OH")
-OH")







